PO.CL01.17 · 临床研究
Validation of microenvironment gene signatures from FFPE to predict nasopharyngeal cancer prognosis
作者与单位 Authors & Affiliations
摘要 Abstract
Background: Nasopharyngeal carcinoma (NPC) is endemic to Southern China and Southeast Asia. Gene expression profiling and biomarker identification in NPC with conventional bulk RNAseq has been challenging due to substantial heterogeneity in cellular composition and limited tissue availability. While we have previously described the microdissected gene expression landscape of NPC and its tumor microenvironment (TME) subtypes, prognostic gene signatures for treatment response remain poorly defined.
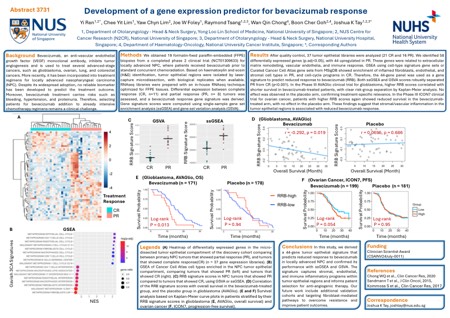
Methods: We obtained formalin-fixed paraffin-embedded (FFPE) biopsies from 64 NPC patients matched for stage but with different clinical outcomes, i.e., the treatment failure group (recurrent/metastatic disease) and the survivor group (remained disease-free). Laser-capture microdissection was performed to isolate the tumor epithelial (TUM) and TME regions based on H&E staining and pathologist annotation. Gene expression libraries were prepared using a specialized RNAseq protocol optimized for FFPE tissues. Following next generation sequencing, bioinformatic analyses were performed to develop gene signatures predictive of clinical outcomes.
Results: After quality control, 277 gene expression libraries were analyzed, consisting of 154 TUM and 123 TME libraries. Unsupervised consensus clustering of TME libraries identified three conserved clusters (average Silhouette width = 0.89). These TME clusters were characterized as immune (C1), epithelial-infiltrative (C2), and stromal (C3) gene signatures based on gene set enrichment analysis (GSEA) and in-silico deconvolution. Notably, 63.2% and 72.7% of TME libraries in C1 and C3 were from survivors, while 61.5% in C2 were from treatment failures (chi-square test, p = 0.0397). Gene signatures for TME clusters were derived from upregulated genes in each cluster, and single-sample GSEA (ssGSEA) was used to calculate signature scores in two independent NPC cohorts (PMID:28851814 and PMID:40412382). Consistently, Kaplan-Meier analyses showed that patients with high C1 or low C2 scores had better progression-free survival (log-rank p-values 0.005 to 0.031). Interestingly, these TME cluster gene signatures outperformed the gene signature derived directly from the treatment failure versus survivor comparison in TME, suggesting that biologically meaningful TME-based gene signatures capture prognostic signal more robustly and generalize better across cohorts.
Conclusions: In this study, we identified three distinct TME clusters in NPC and derived gene signatures that consistently predict progression-free survival across cohorts. These results highlight the value of TME-based biomarkers for risk stratification and precision medicine. Importantly, our FFPE LCM RNAseq workflow enabled high-quality, cell type-specific profiling from archival biopsies, supporting its utility for uncovering clinically relevant biomarkers.
利益披露 Disclosure
Y. Ren, None..
W. Teo, None..
B. Wu, None.
J. W. Foley,
Picopoint Genomics Stock, Patent.
C. Lim, None..
S. Siow, None..
H. Goh, None..
E. Yeo, None..
E. Ong, None..
M. Chua, None..
J. Liu, None..
K. Loh, None..
R. Tsang, None.
J. K. Tay,
Picopoint Genomics Stock, Patent.